


Epigenomes 2024, 8(2), 21; https://doi.org/10.3390/epigenomes8020021 - 27 May 2024
Abstract
Variant H3.3, along with H2A.Z, is notably enriched at promoter regions and is commonly associated with transcriptional activation. However, the specific molecular mechanisms through which H3.3 influences chromatin dynamics at transcription start sites, and its role in gene regulation, remain elusive. Using a
[...] Read more.
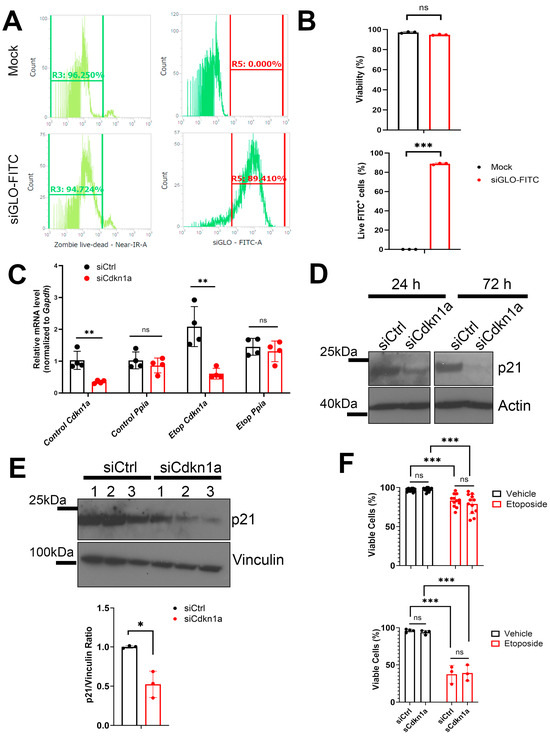
Variant H3.3, along with H2A.Z, is notably enriched at promoter regions and is commonly associated with transcriptional activation. However, the specific molecular mechanisms through which H3.3 influences chromatin dynamics at transcription start sites, and its role in gene regulation, remain elusive. Using a combination of biochemistry and cryo-electron microscopy (cryo-EM), we show that the inclusion of H3.3 alone does not induce discernible changes in nucleosome DNA dynamics. Conversely, the presence of both H3.3 and H2A.Z enhances DNA’s flexibility similarly to H2A.Z alone. Interestingly, our findings suggest that the presence of H3.3 in the H2A.Z nucleosome provides slightly increased protection to DNA at internal sites within the nucleosome. These results imply that while H2A.Z at active promoters promotes the formation of more accessible nucleosomes with increased DNA accessibility to facilitate transcription, the simultaneous presence of H3.3 offers an additional mechanism to fine-tune nucleosome accessibility and the chromatin environment.
Full article
(This article belongs to the Special Issue Histone Variants)
►
Show Figures

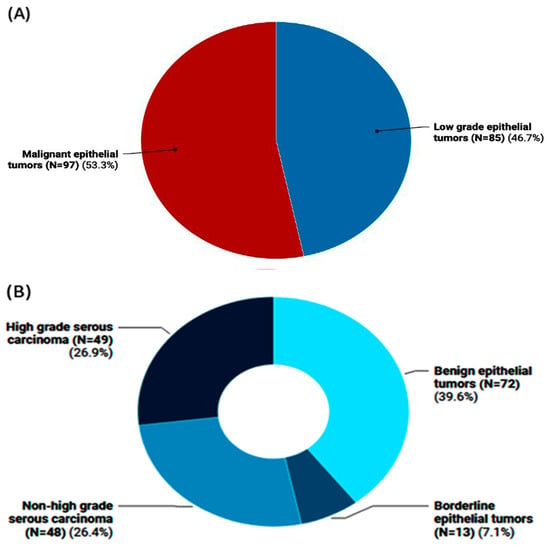
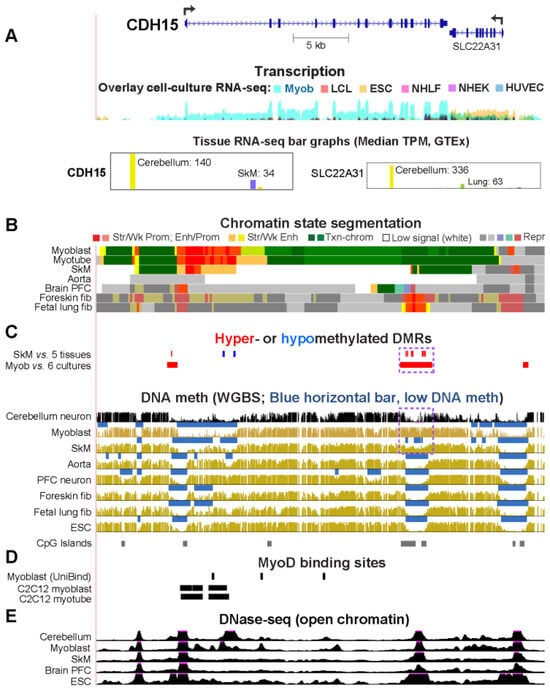
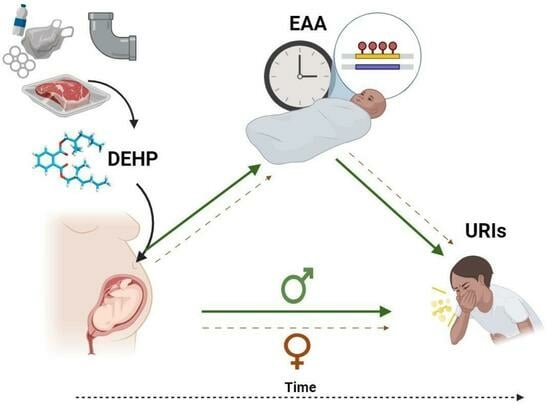
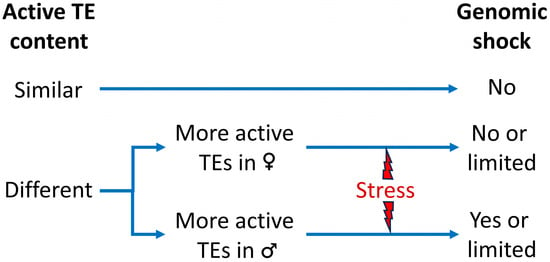
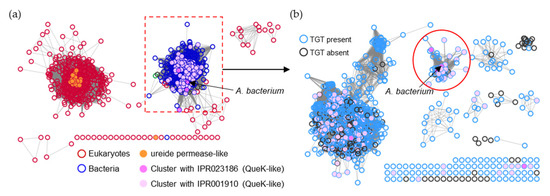
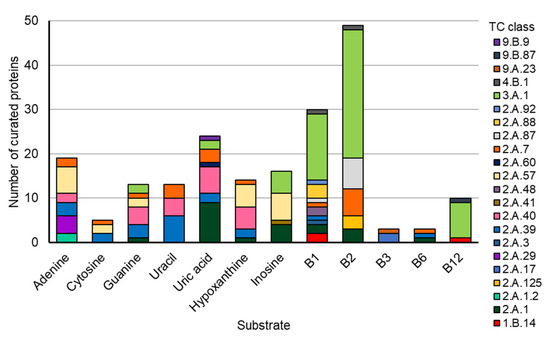
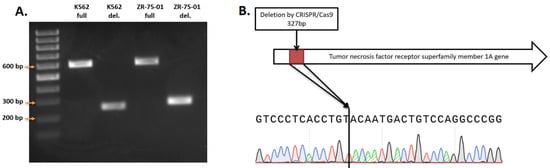
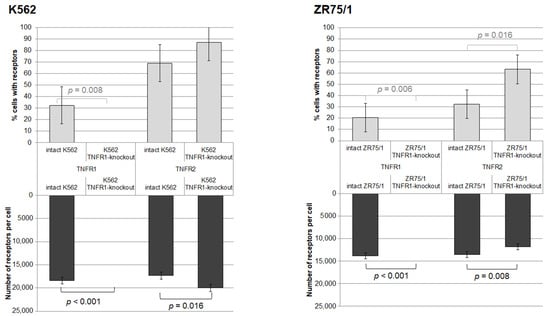
Figure 1



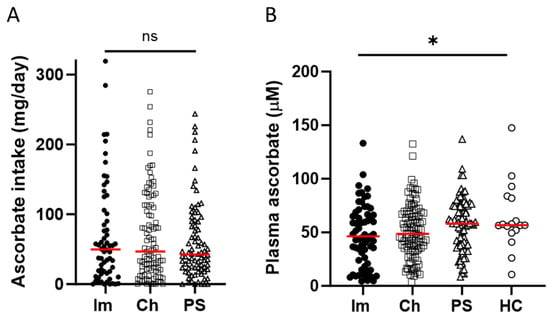






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}